基本信息
- 疾病名称
- 特发性肺纤维化
- 英文名称
- idiopathic pulmonary fibrosis
- 就诊科室
- 呼吸科
- 多发群体
- 50~70岁男性
- 常见症状
- 呼吸困难,咳嗽、咳痰,消瘦、乏力、食欲减退
病因病机
IPF病因不明,发病机制亦未完全阐明,但有足够证据表明与免疫炎症损伤有关。不同标本所显示的免疫炎症反应特征不尽一致,周围血所反映出的是免疫异常比较突出,而支气管肺泡灌洗液显示炎症反应为主,而肺局部组织的异常又有所不同。因此在评估各种研究资料需要考虑到这种差异。目前认为,肺泡上皮细胞损伤和异常修复是导致肺纤维化的主要机制。损伤发生后,修复过程中不能完成正常的再上皮化过程,进而导致肺泡-毛细血管损伤。这一过程诱发细胞因子产生,成纤维细胞表面表达细胞因子受体,在细胞因子作用下聚集到损伤部位并增殖。
临床表现
约15%的IPF病例呈急性,因上呼吸道感染就诊而发现进行性呼吸困难加重,多于6个月内死于呼吸循环衰竭。绝大多数IPF为慢性型(可能尚有介于中间的亚急性型),虽称慢性平均生存时间也只有3.2年。慢性型似乎并非急性型演变而来,确切关系尚不了解。
1.主要症状
(1)呼吸困难劳力性呼吸困难并进行性加重、呼吸浅速可有鼻翼搧动和辅助肌参与呼吸,但大多没有端坐呼吸。
(2)咳嗽、咳痰早期无咳嗽,后可有干咳或少量黏液痰,易有继发感染。出现黏液脓性痰或脓痰,偶见血痰。
(3)全身症状可有消瘦、乏力、食欲不振、关节酸痛等,一般比较少见,急性型可有发热。
2.常见体征
(1)呼吸困难和发绀。
(2)胸廓扩张和膈肌活动度降低。
(3)两肺中下部Velcro啰音,具有一定特征性。
(4)杵状指趾。
(5)终末期呼吸衰竭和右心衰竭相应征象。
诊断及鉴别诊断
1. 临床表现:发病年龄多在中年及以上,男性多于女性。起病隐袭,主要表现为干咳、进行性呼吸困难,活动后明显。多数患者双下肺可闻及吸气末爆裂音或捻发音,超过半数可见杵状指(趾)。终末期出现紫绀、肺动脉高压、肺心病和右心功能不全。
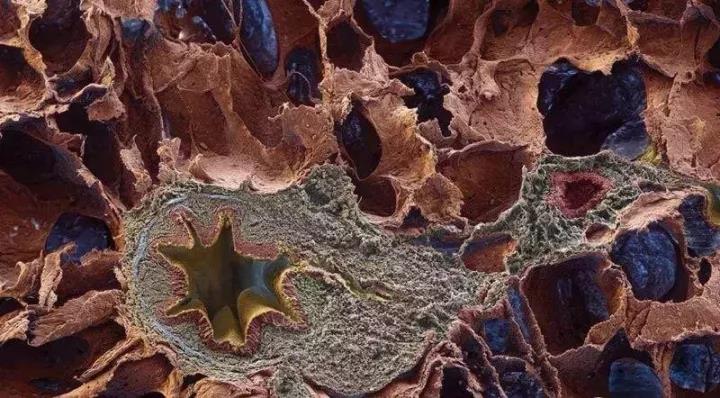
2. 胸部HRCT:胸部HRCT是诊断IPF的必要诊断手段。胸片对于疑似IPF患者的诊断价值明显地低于HRCT,不应该作为IPF的诊断依据。IPF的胸部HRCT以网格影和蜂窝肺为主要特征,经常伴有牵拉性支气管扩张[2],其中蜂窝肺是诊断肯定UIP型重要的依据。少见磨玻璃影,且通常比网格影范围小。胸部HRCT典型的UIP分布特征是以双下肺和外带分布。
合并胸膜异常,如胸膜斑、钙化、显著的胸腔积液时,多提示为导致UIP型病变由其他疾病引起。微结节、气体陷闭、非蜂窝肺的囊腔影、广泛的磨玻璃影、实变,或沿支气管血管为著的分布特点,均提示其他的诊断。
3. 组织病理学检查:IPF的特征性组织病理学改变是普通型间质性肺炎,其主要病变为纤维化,病变的程度及分布不一致,低倍显微镜下观察,同时可见瘢痕纤维化区域伴有蜂窝肺改变,和病变较轻甚至正常肺组织区域[2]。这些组织病理改变通常以胸膜下和间隔旁肺实质为著。通常炎症较为轻微,可有少量淋巴细胞和浆细胞间质浸润,伴2型肺泡上皮细胞和细支气管上皮细胞增生。纤维化区域主要由致密的胶原纤维组成,可见散在分布的成纤维细胞灶。蜂窝肺区域由囊性纤维化的气腔组成,通常衬附着细支气管上皮细胞,腔内有粘液和炎症细胞填充。肺纤维化区域和蜂窝肺病变区域中,肺脏间质可见平滑肌增生。
4. 诊断标准
(1)除外已知病因所致的间质性肺疾病,如职业接触、室内外环境暴露、结缔组织病和药物性肺损害等。
(2)未行外科肺活检的患者,胸部HRCT表现为肯定UIP型。
(3)行外科肺活检的患者,结合HRCT和外科肺活检符合特定的类型。
治疗及预后
(一)药物治疗
(1)吡非尼酮:
吡非尼酮是2015年美国胸科学会/欧洲呼吸协会/日本胸科协会/拉丁美洲胸科协会联合发布的《特发性肺纤维化临床治疗推荐指南》中个推荐用于治疗的两个药物之一, IPF患者经吡非尼酮连续服药治疗52周后,能够减缓患者的肺功能指标如FVC和DLCO的下降,延长患者无进展生存期(PFS),同时降低患者死亡风险。
(2)尼达尼布:尼达尼布是2015年美国胸科学会/欧洲呼吸协会/日本胸科协会/拉丁美洲胸科协会联合发布的《特发性肺纤维化临床治疗推荐指南》中推荐等级最高(条件推荐)的两大药物之一。
(二)非药物治疗
① 戒烟:大多数IPF患者是吸烟者,吸烟与疾病的发生具有一定的相关性。吸烟者,必须劝导和帮助患者戒烟。②氧疗。③机械通气。④肺康复。⑤肺移植。
康安途医疗旅游 时间:2018-09-15 所属标签:吡非尼酮,PIRFENIDONE
- 词条统计
-
- 浏览次数: 1752次
- 最近更新: 2018-08-29
- 创建者: 新垣结衣






不适用于哪些非小细胞肺癌患者")








